Workflow templates and sample data
Note: the most recent version of this vignette can be found here.
Note: if you use
systemPipeRorsystemPipeRdatain published research, please cite: Backman, T.W.H and Girke, T. (2016). systemPipeR: Workflow and Report Generation Environment. BMC Bioinformatics, 17: 388. 10.1186/s12859-016-1241-0.
systemPipeRdata package provides a demo sample FASTQ files used in the
workflow reporting vignettes. The chosen data set SRP010938 obtains 18
paired-end (PE) read sets from Arabidposis thaliana (Howard et al. 2013). To
minimize processing time during testing, each FASTQ file has been subsetted to
90,000-100,000 randomly sampled PE reads that map to the first 100,000
nucleotides of each chromosome of the A. thalina genome. The corresponding
reference genome sequence (FASTA) and its GFF annotation files (provided in the
same download) have been truncated accordingly. This way the entire test sample
data set requires less than 200MB disk storage space. A PE read set has been
chosen for this test data set for flexibility, because it can be used for
testing both types of analysis routines requiring either SE (single-end) reads
or PE reads.
Getting started
Installation
The systemPipeRdata package is available at Bioconductor and can be installed from within R as follows:
if (!requireNamespace("BiocManager", quietly = TRUE)) install.packages("BiocManager")
BiocManager::install("systemPipeRdata")
Also, it is possible to install the development version from Bioconductor.
BiocManager::install("systemPipeRdata", version = "devel", build_vignettes = TRUE,
dependencies = TRUE) # Installs Devel version from Bioconductor
Loading package and documentation
library("systemPipeRdata") # Loads the package
library(help = "systemPipeRdata") # Lists package info
vignette("systemPipeRdata") # Opens vignette
Starting with pre-configured workflow templates
Load one of the available workflows into your current working directory.
The following does this for the rnaseq workflow template. The name of the resulting
workflow directory can be specified under the mydirname argument. The default NULL
uses the name of the chosen workflow. An error is issued if a directory of the same
name and path exists already.
Full details of pre-configed workflows are listed on this page
genWorkenvir(workflow = "systemPipeR/SPrnaseq", mydirname = "rnaseq")
setwd("rnaseq")
On Linux and OS X systems the same can be achieved from the command-line of a terminal with the following commands.
$ Rscript -e "systemPipeRdata::genWorkenvir(workflow='systemPipeR/SPrnaseq', mydirname='rnaseq')"
Build, run and visualize the workflow template
- Build workflow from RMarkdown file
This template provides some common steps for a RNAseq workflow. One can add, remove, modify
workflow steps by operating on the sal object.
sal <- SPRproject()
sal <- importWF(sal, file_path = "systemPipeVARseq.Rmd", verbose = FALSE)
- Running workflow
Next, we can run the entire workflow from R with one command:
sal <- runWF(sal)
- Visualize workflow
systemPipeR workflows instances can be visualized with the plotWF function.
plotWF(sal)
- Report generation
systemPipeR compiles all the workflow execution logs in one central location,
making it easier to check any standard output (stdout) or standard error
(stderr) for any command-line tools used on the workflow or the R code stdout.
sal <- renderLogs(sal)
Also, the technical report can be generated using renderReport function.
sal <- renderReport(sal)
Workflow templates collection
A collection of workflow templates are available, and it is possible to browse the current availability, as follows:
availableWF(github = TRUE)
This function returns the list of workflow templates available within the package and systemPipeR Organization on GitHub. Each one listed template can be created as described above.
The workflow template choose from Github will be installed as an R package, and also it creates the environment with all the settings and files to run the demo analysis.
genWorkenvir(workflow="systemPipeR/SPrnaseq", mydirname="NULL")
setwd("SPrnaseq")
Besides, it is possible to choose different versions of the workflow template,
defined through other branches on the GitHub Repository. By default, the master
branch is selected, however, it is possible to define a different branch with the ref argument.
genWorkenvir(workflow="systemPipeR/SPrnaseq", ref = "singleMachine")
setwd("SPrnaseq")
Download a specific R Markdown file
Also, it is possible to download a specific workflow script for your analysis.
The URL can be specified under url argument and the R Markdown file name in
the urlname argument. The default NULL copies the current version available in the chose template.
genWorkenvir(workflow="systemPipeR/SPrnaseq", url = "https://raw.githubusercontent.com/systemPipeR/systemPipeRNAseq/cluster/vignettes/systemPipeRNAseq.Rmd",
urlname = "rnaseq_V-cluster.Rmd")
setwd("rnaseq")
Dynamic generation of workflow template
It is possible to create a new workflow structure from RStudio
menu File -> New File -> R Markdown -> From Template -> systemPipeR New WorkFlow.
This interactive option creates the same environment as demonstrated above.
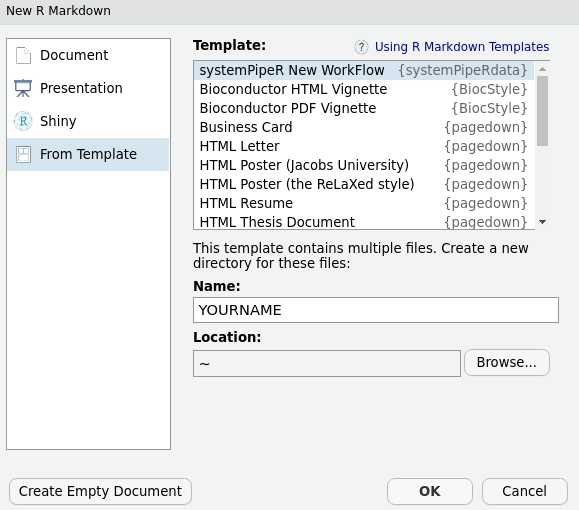 Figure 1: Selecting workflow template within RStudio.
Figure 1: Selecting workflow template within RStudio.
Directory Structure
The workflow templates generated by genWorkenvir contain the following preconfigured directory structure:
- workflow/ (e.g. rnaseq/)
- This is the root directory of the R session running the workflow.
- Run script ( *.Rmd) and sample annotation (targets.txt) files are located here.
- Note, this directory can have any name (e.g. rnaseq, varseq). Changing its name does not require any modifications in the run script(s).
- Important subdirectories:
- param/
- Stores non-CWL parameter files such as: *.param, *.tmpl and *.run.sh. These files are only required for backwards compatibility to run old workflows using the previous custom command-line interface.
- param/cwl/: This subdirectory stores all the CWL parameter files. To organize workflows, each can have its own subdirectory, where all
CWL paramandinput.ymlfiles need to be in the same subdirectory.
- data/
- FASTQ files
- FASTA file of reference (e.g. reference genome)
- Annotation files
- etc.
- results/
- Analysis results are usually written to this directory, including: alignment, variant and peak files (BAM, VCF, BED); tabular result files; and image/plot files
- Note, the user has the option to organize results files for a given sample and analysis step in a separate subdirectory.
- param/
Note: Directory names are indicated in green. Users can change this structure as needed, but need to adjust the code in their workflows accordingly.
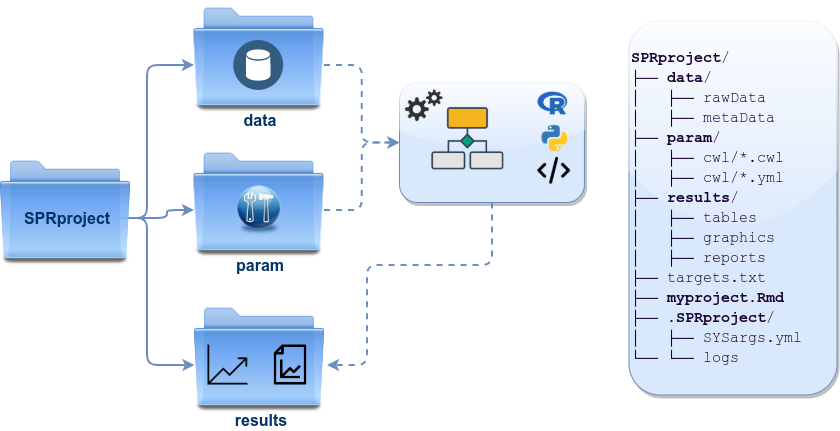
Figure 2: systemPipeR’s preconfigured directory structure.
Return paths to sample data
The location of the sample data provided by systemPipeRdata can be returned as a list.
pathList()[1:2]
## $targets
## [1] "C:/Users/lz/AppData/Local/R/win-library/4.2/systemPipeRdata/extdata/param/targets.txt"
##
## $targetsPE
## [1] "C:/Users/lz/AppData/Local/R/win-library/4.2/systemPipeRdata/extdata/param/targetsPE.txt"
Version information
sessionInfo()
## R version 4.2.0 (2022-04-22 ucrt)
## Platform: x86_64-w64-mingw32/x64 (64-bit)
## Running under: Windows 10 x64 (build 19044)
##
## Matrix products: default
##
## locale:
## [1] LC_COLLATE=English_United States.utf8
## [2] LC_CTYPE=English_United States.utf8
## [3] LC_MONETARY=English_United States.utf8
## [4] LC_NUMERIC=C
## [5] LC_TIME=English_United States.utf8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets
## [6] methods base
##
## other attached packages:
## [1] systemPipeRdata_2.1.1 BiocStyle_2.24.0
##
## loaded via a namespace (and not attached):
## [1] bslib_0.3.1 compiler_4.2.0
## [3] BiocManager_1.30.18 formatR_1.12
## [5] jquerylib_0.1.4 GenomeInfoDb_1.32.1
## [7] XVector_0.36.0 bitops_1.0-7
## [9] remotes_2.4.2 tools_4.2.0
## [11] zlibbioc_1.42.0 digest_0.6.29
## [13] jsonlite_1.8.0 evaluate_0.15
## [15] rlang_1.0.2 cli_3.3.0
## [17] rstudioapi_0.13 yaml_2.3.5
## [19] blogdown_1.10.1 xfun_0.31
## [21] fastmap_1.1.0 GenomeInfoDbData_1.2.8
## [23] stringr_1.4.0 knitr_1.39
## [25] Biostrings_2.64.0 sass_0.4.1
## [27] S4Vectors_0.34.0 IRanges_2.30.0
## [29] stats4_4.2.0 R6_2.5.1
## [31] rmarkdown_2.14 bookdown_0.26
## [33] magrittr_2.0.3 codetools_0.2-18
## [35] htmltools_0.5.2 BiocGenerics_0.42.0
## [37] stringi_1.7.6 RCurl_1.98-1.6
## [39] crayon_1.5.1
Funding
This project was supported by funds from the National Institutes of Health (NIH) and the National Science Foundation (NSF).
References
Howard, Brian E, Qiwen Hu, Ahmet Can Babaoglu, Manan Chandra, Monica Borghi, Xiaoping Tan, Luyan He, et al. 2013. “High-Throughput RNA Sequencing of Pseudomonas-Infected Arabidopsis Reveals Hidden Transcriptome Complexity and Novel Splice Variants.” PLoS One 8 (10): e74183. https://doi.org/10.1371/journal.pone.0074183.