systemPipeR: Project structure
Last update: 06 August, 2021
spr_project.RmdAuthors: Daniela Cassol (danielac@ucr.edu), Le Zhang (le.zhang001@email.ucr.edu), Thomas Girke (thomas.girke@ucr.edu).
Institution: Institute for Integrative Genome Biology, University of California, Riverside, California, USA.
Project structure
systemPipeR expects a project directory structure that consists of a directory where users may store all the raw data (e.g. data/) , the results directory that will be reserved for all the outfiles files (e.g. results/) , and the parameters directory (e.g. param/).
This structure allows reproducibility and collaboration across the data science team, since internally relative paths are used. Users could transfer the project to a different location and still be able to run the entire workflow. Also, it increases efficiency and data management once the raw data is kept in a separate folder and avoids duplication.
Directory Structure
systemPipeRdata, helper package, provides pre-configured workflows, reporting templates, and sample data loaded as demonstrated below. With a single command, the package allows creating the workflow environment containing the structure described here (see Figure 1).
Directory names are indicated in green. Users can change this structure as needed, but need to adjust the code in their workflows accordingly.
- workflow/ (e.g. myproject/)
- This is the root directory of the R session running the workflow.
- Run script ( *.Rmd) and sample annotation (targets.txt) files are located here.
- Note, this directory can have any name (e.g. myproject). Changing its name does not require any modifications in the run script(s).
-
Important subdirectories:
- param/
-
param/cwl/: This subdirectory stores all the parameter and configuration files. To organize workflows, each can have its own subdirectory, where all
*.cwland*input.ymlfiles need to be in the same subdirectory.
-
data/
- Raw data (e.g. FASTQ files)
- FASTA file of reference (e.g. reference genome)
- Annotation files
- Metadata
- etc.
-
results/
- Analysis results are usually written to this directory, including: alignment, variant and peak files (BAM, VCF, BED); tabular result files; and image/plot files
- Note, the user has the option to organize results files for a given sample and analysis step in a separate subdirectory.
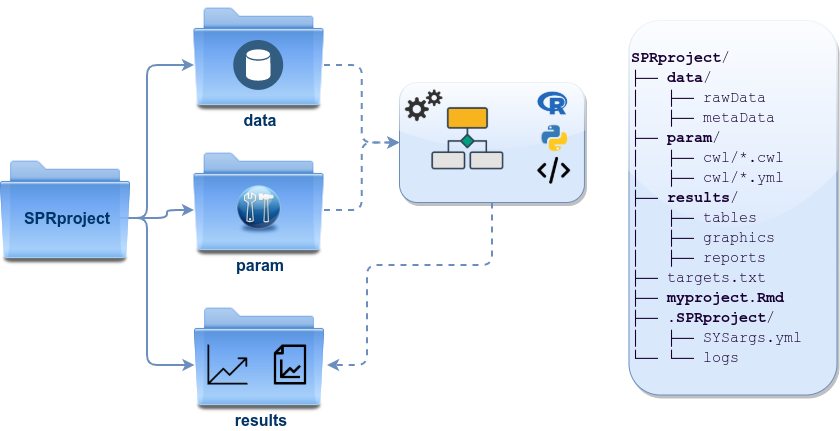
Figure 1. systemPipeR’s preconfigured directory structure.
Structure of initial targets file
The targets file defines all input files (e.g. FASTQ, BAM, BCF) and sample comparisons of an analysis workflow. The following shows the format of a sample targets file included in the package. It also can be viewed and downloaded from systemPipeR’s GitHub repository here. In a target file with a single type of input files, here FASTQ files of single-end (SE) reads, the first column defines the paths and the second column represents a unique id name for each sample. The third column called Factor represents the biological replicates. All subsequent columns are optional to provide additional information. Any number of additional columns can be added as needed.
Users should note here, the usage of targets files is optional when using systemPipeR's new workflow management interface. They can be replaced by a standard YAML input file used by CWL. Since for organizing experimental variables targets files are extremely useful and user-friendly. Thus, we encourage users to keep using them.
- Structure of
targetsfile for paired-end (PE) samples
targetspath <- system.file("extdata", "targetsPE.txt", package="systemPipeR")
showDF(read.delim(targetspath, comment.char = "#"))To work with custom data, users need to generate a targets file containing the paths to their own FASTQ files and then provide under targetspath the path to the corresponding targets file.