layout: true background-image: url(https://raw.githubusercontent.com/tgirke/systemPipeR/gh-pages/images/systemPipeR.png) background-position: 99% 1% background-size: 10%
{kind=link}
class: middle
Outline
Introduction
Design
How to run a Workflow
Workflows Tutorial
Live Demo
class: inverse, center, middle
Introduction
Introduction
systemPipeR provides a suite of R/Bioconductor packages for designing, building and running end-to-end analysis workflows on local machines, HPC clusters and cloud systems, while generating at the same time publication quality analysis reports
systemPipeR offers many utilities to build, control, and execute workflows entirely from R
The environment takes advantage of central community S4 classes of the Bioconductor ecosystem
Workflows are managed by generic workflow management containers supporting both analysis routines implemented in R code and/or command-line software
Simple annotation system targets
systemPipeR’s Core Functionalities
.center[
 ]
]
Structural Features
– .left-column[
WF infrastructure
]
.right-column[ systemPipeR offers many utilities to build, control, and execute workflows entirely from R. The environment takes advantage of central community S4 classes of the Bioconductor ecosystem. Workflows are managed by generic workflow management containers supporting both analysis routines implemented in R code and/or command-line software. A layered monitoring infrastructure is provided to design, control and debug each step in a workflow. The run environment allows to execute workflows entirely or with a intuitive to use step-wise execution syntax using R’s standard subsetting syntax (runWF(sys[1:3])) or pipes (%>%). ]
Structural Features
.left-column[
WF infrastructure
Command-line support
]
.right-column[ An important feature of systemPipeR is support for running command-line software by adopting the Common Workflow Language (CWL). The latter is a widely adopted community standard for describing analysis workflows. This design offers several advantages such as:
seamless integration of most command-line software
support to run systemPipeR workflows from R or many other popular computer languages
efficient sharing of workflows across different workflow environments. ]
Structural Features
.left-column[
WF infrastructure
Command-line support
Parallel evaluation
]
.right-column[ The processing time of workflows can be greatly reduced by making use of parallel evaluations across several CPU cores on single machines, or multiple nodes of computer clusters and cloud-based systems. systemPipeR simplifies these parallelization tasks without creating any limitations for users who do not have access to high-performance computer resources ]
Structural Features
.left-column[
WF infrastructure
Command-line support
Parallel evaluation
Reports infrastructure
]
.right-column[ systemPipeR’s reporting infrastructure includes three types of interconnected reports each serving a different purpose:
a scientific report, based on R Markdown, contains all scientifically relevant results
a technical report captures all technical information important for each workflow step, including parameter settings, software versions, and warning/error messages, etc.
a visual report depicts the entire workflow topology including its run status in form of a workflow graph
]
Structural Features
.left-column[
WF infrastructure
Command-line support
Parallel evaluation
Reports infrastructure
Shiny Web Interface
]
.right-column[ Recently, the systemPipeShiny package has been added that allows users to design workflows in an interactive graphical user interface (GUI). In addition to designing workflows, this new interface allows users to run and to monitor workflows in an intuitive manner without the need of knowing R. ]
Structural Features
.left-column[
WF infrastructure
Command-line support
Parallel evaluation
Reports infrastructure
Shiny Web Interface
Workflow Templates
]
.right-column[ A rich set of end-to-end workflow templates is provided by this project for a wide range omics applications. In addition, users can contribute and share their workflows with the community by submitting them to a central GitHub repository ]
Important Functions
.small[
| Function Name | Description | Category |
|---|---|---|
genWorkenvir |
Generates workflow templates provided by systemPipeRdata helper package / or from the individuals’ pipelines packages | Accessory |
loadWorkflow |
Constructs SYSargs2 object from CWL param and targets files |
SYSargs2 |
renderWF |
Populate all the command-line in an SYSargs2 object |
SYSargs2 |
subsetWF |
Subsetting SYSargs2 class slots |
SYSargs2 |
runCommandline |
Executes command-line software on samples and parameters specified in SYSargs2 object |
SYSargs2 |
clusterRun |
Runs command-line software in parallel mode on a computer cluster | SYSargs2 |
writeTargetsout |
Write updated targets out to file/Generate targets file with reference | SYSargs2 |
output_update |
Updates the output files paths in the SYSargs2 object |
SYSargs2 |
singleYML |
Create automatically the param.yml |
SYSargs2 |
createWF |
Create automatically param.cwl and the param.yml based on the command line |
SYSargs2 |
config.param |
Custom configuration of the CWL param files from R | SYSargs2 |
| ] |
Important Functions
.small[
| Function Name | Description | Category |
|---|---|---|
initWF |
Constructs SYSargsList workflow control module (S4 object) from script file |
SYSargsList |
configWF |
Control of which step of the workflow will be run and the generation of the new RMarkdown | SYSargsList |
runWF |
Runs all the R chunk define in the RMarkdown file or a subset, e.g. runWF[1:3] |
SYSargsList |
renderReport |
Render Scientific Report based on RMarkdown | SYSargsList |
subsetRmd |
Write updated subset Rmarkdown of R chunk with text associate in the step selected | SYSargsList |
plotWF |
Plot visual workflow designs and topologies with different graphical layouts | SYSargsList |
statusWF |
Return the overview of the workflow steps computational status | SYSargsList |
evalCode |
Turn eval option TRUE or FALSE on RMarkdown file |
Accessory |
tryCL |
Checks if third-party software or utility is installed and set in the PATH | Accessory |
| ] |
class: inverse, center, middle
Design
Workflow Management Solutions
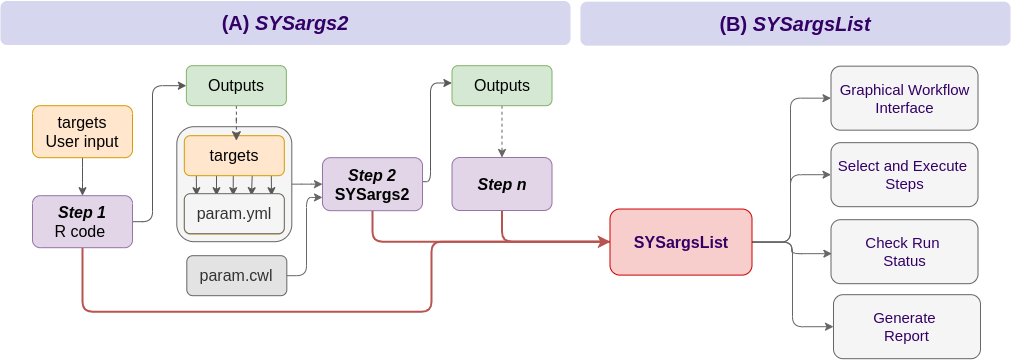
systemPipeR central concept for designing workflows is workflow management containers (S4 class)
SYSargs2 controls workflow steps with input/output file operations
SYSargs2 requires a targets and a set of workflow definition files (here param.cwl and param.yml)
SYSargsList objects organize one or many SYSargs2 containers in a single compound object capturing all information required to run, control and monitor complex workflows from start to finish
.center[
 ]
]
Directory Structure
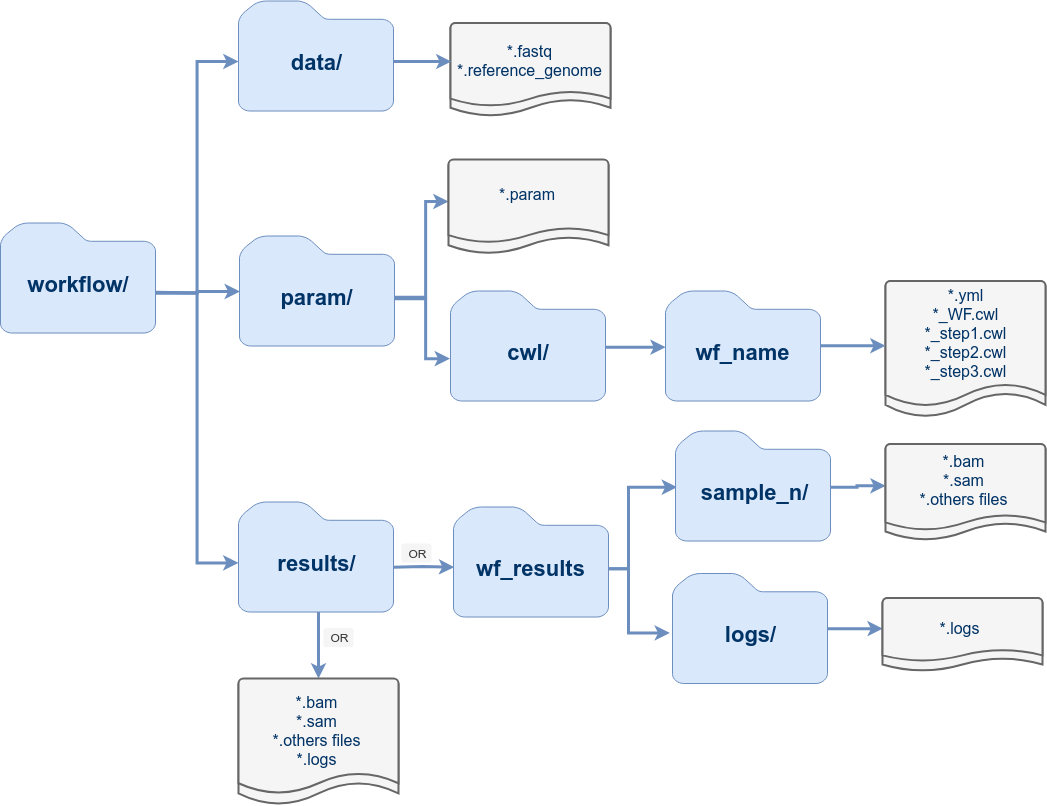
The workflow templates generated by genWorkenvir contain the following preconfigured directory structure:
Workflows Collection
Browse pipelines that are currently available as part of the systemPipeR toolkit
.small[
| WorkFlow | Description | Version | GitHub | CI Testing |
|---|---|---|---|---|
| systemPipeChIPseq | ChIP-Seq Workflow Template | v1.0  |
 |
|
| systemPipeRIBOseq | RIBO-Seq Workflow Template | v1.0 |
|
|
| systemPipeRNAseq | RNA-Seq Workflow Template | v1.0 |
|
|
| systemPipeVARseq | VAR-Seq Workflow Template | v1.0 |
|
|
| systemPipeMethylseq | Methyl-Seq Workflow Template | devel  |
|
|
| systemPipeDeNovo | De novo transcriptome assembly Workflow Template | devel |
|
|
| systemPipeCLIPseq | CLIP-Seq Workflow Template | devel |
|
|
| systemPipeMetaTrans | Metatranscriptomic Sequencing Workflow Template | devel |
|
|
| ] |
class: inverse, center, middle
CWL
CWL
TODO: Add section with CWL details
.center[
 ]
]
CWL and SPR
TODO: How to use CWL definition with systemPipeR
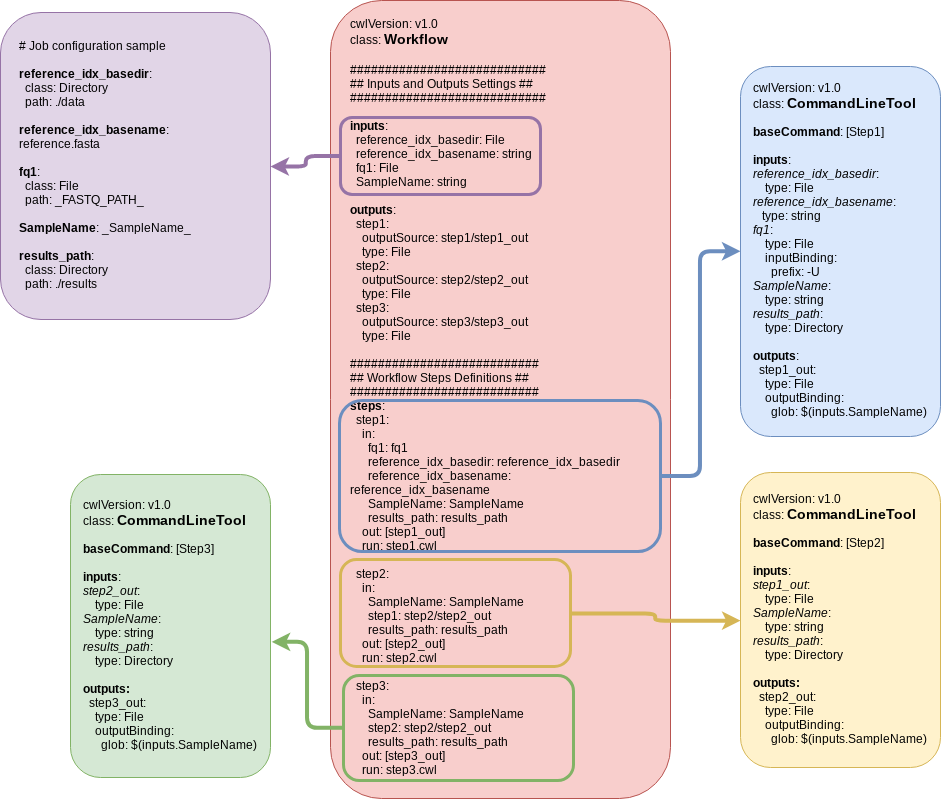
- SYSargs2 instances are constructed from a
targetsfile and twoparamfilehisat2-mapping-se.cwlfile contains the settings for running command-line softwarehisat2-mapping-se.ymlfile define all the variables to be input in the specific command-line step
targets <- system.file("extdata", "targets.txt", package="systemPipeR")
dir_path <- system.file("extdata/cwl/hisat2/hisat2-se", package="systemPipeR")
align <- loadWF(targets=targets, wf_file="hisat2-mapping-se.cwl",
input_file="hisat2-mapping-se.yml", dir_path=dir_path)
align <- renderWF(align, inputvars=c(FileName="_FASTQ_PATH_", SampleName="_SampleName_"))
## Instance of 'SYSargs2':
## Slot names/accessors:
## targets: 18 (M1A...V12B), targetsheader: 4 (lines)
## modules: 2
## wf: 0, clt: 1, yamlinput: 7 (components)
## input: 18, output: 18
## cmdlist: 18
## WF Steps:
## 1. hisat2-mapping-se.cwl (rendered: TRUE)
CWL and SPR
SYSargs2 instance
- Slots and accessor functions have the same names
names(align)
# [1] "targets" "targetsheader" "modules" "wf" "clt"
# [6] "yamlinput" "cmdlist" "input" "output" "cwlfiles"
# [11] "inputvars"
cmdlistreturn command-line arguments for the specific software, hereHISAT2for the first sample
cmdlist(align)[1]
# $M1A
# $M1A$`hisat2-mapping-se.cwl`
# [1] "hisat2 -S results/M1A.sam -x ./data/tair10.fasta -k 1 --min-intronlen 30 --max-intronlen 3000 -U ./data/SRR446027_1.fastq.gz --threads 4"
- The output components of
SYSargs2define all the expected output files for each step in the workflow; some of which are the input for the next workflow step
output(align)[1]
# $M1A
# $M1A$`hisat2-mapping-se.cwl`
# [1] "results/M1A.sam"
class: inverse, center, middle
Metadata
Targets file organizes samples
- Structure of
targetsfile for single-end (SE) library
targetspath <- system.file("extdata", "targets.txt", package="systemPipeR")
read.delim(targetspath, comment.char = "#")[1:3,1:4]
## FileName SampleName Factor SampleLong
## 1 ./data/SRR446027_1.fastq.gz M1A M1 Mock.1h.A
## 2 ./data/SRR446028_1.fastq.gz M1B M1 Mock.1h.B
## 3 ./data/SRR446029_1.fastq.gz A1A A1 Avr.1h.A
- Structure of
targetsfile for paired-end (PE) library
targetspath <- system.file("extdata", "targetsPE.txt", package="systemPipeR")
read.delim(targetspath, comment.char = "#")[1:3,1:5]
## FileName1 FileName2 SampleName Factor
## 1 ./data/SRR446027_1.fastq.gz ./data/SRR446027_2.fastq.gz M1A M1
## 2 ./data/SRR446028_1.fastq.gz ./data/SRR446028_2.fastq.gz M1B M1
## 3 ./data/SRR446029_1.fastq.gz ./data/SRR446029_2.fastq.gz A1A A1
## SampleLong
## 1 Mock.1h.A
## 2 Mock.1h.B
## 3 Avr.1h.A
Integration with SummarizedExperiment
- Integrates targets files and count table from systemPipeR to a SummarizedExperiment object
## Create an object with targets file and comparison and count table
sprSE <- SPRdata(targetspath = targetspath, cmp=TRUE)
metadata(sprSE)
# $version
# [1] ‘1.23.9’
#
# $comparison
# $comparison$CMPset1
# [,1] [,2]
# [1,] "M1" "A1"
# [2,] "M1" "V1"
# [3,] "A1" "V1"
# [4,] "M6" "A6"
colData(sprSE)
# DataFrame with 18 rows and 6 columns
# FileName SampleName Factor SampleLong
# <character> <character> <character> <character>
# M1A ./data/SRR446027_1.f.. M1A M1 Mock.1h.A
# M1B ./data/SRR446028_1.f.. M1B M1 Mock.1h.B
# ... ... ... ... ...
# M12B ./data/SRR446040_1.f.. M12B M12 Mock.12h.B
class: inverse, center, middle
Live Demo
Install Package
Install the systemPipeRdata package from Bioconductor:
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("systemPipeR")
Load Package and Documentation
Load package:
library("systemPipeR")
Access help:
library(help="systemPipeR")
vignette("systemPipeR")
Quick Start
Load Sample Workflow
systemPipeRdata
- Helper package to generate with a single command workflow templates for systemPipeR
- Includes sample data for testing
- User can create new workflows or change and extend existing ones
- Template Workflows:
- Sample workflows can be loaded with the
genWorkenvirfunction from systemPipeRdata
- Sample workflows can be loaded with the
Generate workflow template:
library(systemPipeRdata)
genWorkenvir(workflow="rnaseq")
setwd("rnaseq")
More details about systemPipeRdata package here
Install Workflow
Check the workflow template availability
availableWF(github = TRUE)
# $systemPipeRdata
# [1] "chipseq" "new" "riboseq" "rnaseq" "varseq"
#
# $github
# workflow branches version html description
# 1 systemPipeR/systemPipeChIPseq master release https://github.com/systemPipeR/systemPipeChIPseq Workflow Template
# 2 systemPipeR/systemPipeRIBOseq master release https://github.com/systemPipeR/systemPipeRIBOseq Workflow Template
# 3 systemPipeR/systemPipeRNAseq cluster, master, singleMachine release https://github.com/systemPipeR/systemPipeRNAseq Workflow Template
# 4 systemPipeR/systemPipeVARseq master release https://github.com/systemPipeR/systemPipeVARseq Workflow Template
# 5 systemPipeR/systemPipeCLIPseq master devel https://github.com/systemPipeR/systemPipeCLIPseq Workflow Template
# 6 systemPipeR/systemPipeDeNovo master devel https://github.com/systemPipeR/systemPipeDeNovo Workflow Template
# 7 systemPipeR/systemPipeMetaTrans master devel https://github.com/systemPipeR/systemPipeMetaTrans Workflow Template
# 8 systemPipeR/systemPipeMethylseq master devel https://github.com/systemPipeR/systemPipeMethylseq Workflow Template
Dynamic Workflow Template
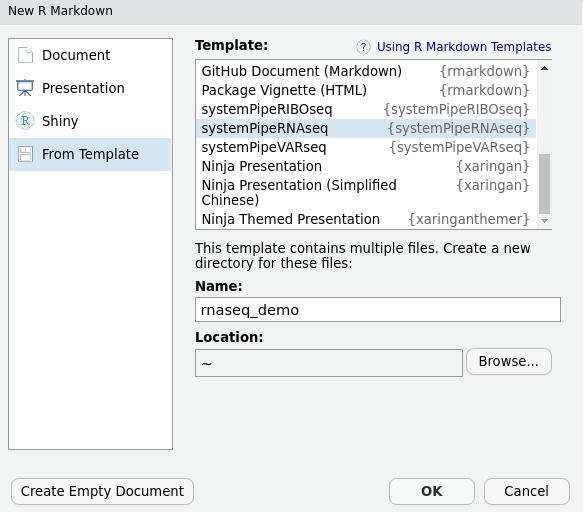
Create dynamic Workflow Templates with RStudio
File -> New File -> R Markdown -> From Template
.center[
 ]
]
Run a Workflow
.left-column[
Setup
]
.right-column[
library(systemPipeR)
targetspath <- system.file("extdata", "targets.txt", package="systemPipeR")
read.delim(targetspath, comment.char = "#")[1:4,1:4]
## FileName SampleName Factor SampleLong
## 1 ./data/SRR446027_1.fastq.gz M1A M1 Mock.1h.A
## 2 ./data/SRR446028_1.fastq.gz M1B M1 Mock.1h.B
## 3 ./data/SRR446029_1.fastq.gz A1A A1 Avr.1h.A
## 4 ./data/SRR446030_1.fastq.gz A1B A1 Avr.1h.B
script <- system.file("extdata/workflows/rnaseq", "systemPipeRNAseq.Rmd", package="systemPipeRdata")
]
Run a Workflow
.left-column[
Setup
initWF
]
.right-column[
sysargslist <- initWF(script = script, targets = targetspath, overwrite = TRUE)
# Project started with success: ./SYSproject and SYSconfig.yml were created.
]
Run a Workflow
.left-column[
Setup
initWF
configWF
]
.right-column[
sysargslist <- configWF(sysargslist, input_steps = "1:3")
sysargslist
# Instance of 'SYSargsList':
# WF Steps:
# 1. Rmarkdown/HTML setting
# 2. Introduction
# 3. Samples and environment settings
# 3.1. Environment settings and input data
# 3.2. Required packages and resources
# 3.3. Experiment definition provided by `targets` file
]
Run a Workflow
.left-column[
Setup
initWF
configWF
runWF
]
.right-column[
sysargslist <- runWF(sysargslist, steps = "1:2")
# Step: 1: Introduction --> DONE
# Step: 2: Samples and environment settings --> DONE
# Step: 2.1: Environment settings and input data --> DONE
# Step: 2.2: Required packages and resources --> DONE
# Step: 2.3: Experiment definition provided by `targets` file --> DONE
sysargslist <- runWF(sysargslist, steps = "ALL")
]
Run a Workflow
.left-column[
Setup
initWF
configWF
runWF
renderReport
]
.right-column[
sysargslist <- renderReport(sysargslist = sysargslist)
]
How to Use Pipes %>%
Consider the following example, in which the steps are the initialization, configuration and running the entire workflow.
library(systemPipeR)
sysargslist <- initWF(script ="systemPipeRNAseq.Rmd", overwrite = TRUE) %>%
configWF(input_steps = "1:6") %>%
runWF(steps = "1:2")
class: inverse, center, middle
Project Updates
targets x SummarizedExperiment
Extension “SummarizedExperiment” methods:
sprSE <- addAssay(sprSE, assay(countMatrix), xName="countMatrix")
sprSE <- addMetadata(sprSE, list(targets), xName="metadata")
New Function:
## Create empty SummarizedExperiment
sprSE <- SPRdata()
## Create an object with targets file and comparison and count table
sprSE <- SPRdata(counts = countMatrix, cmp=TRUE, targetspath = targetspath)
metadata(sprSE)
colData(sprSE)
assays(sprSE)
SPR Paper
Added the main points to discuss in the draft
Writing: Results and introduction
Improve Graphical Abstract
Show case?
SYSargsList
Explain how SYSargsList is implemented - Vignette
.small[
| Function Name | Description |
|---|---|
initWF |
Constructs SYSargsList workflow control module (S4 object) from script file |
configWF |
Control of which step of the workflow will be run and the generation of the new RMarkdown |
runWF |
Runs all the R chunk define in the RMarkdown file or a subset, e.g. runWF[1:3] |
renderReport |
Render Scientific Report based on RMarkdown |
renderLog |
Render logs Report based on RMarkdown |
updateWF |
Recover the SYSargsList workflow previous ran and restarts the WF |
plotWF |
Plot visual workflow designs and topologies with different graphical layouts |
statusWF |
Return the overview of the workflow steps computational status |
evalCode |
Turn eval option TRUE or FALSE on RMarkdown file |
tryCL |
Checks if third-party software or utility is installed and set in the PATH |
| ] |
Improve statusWF()
Visualization in systemPipeR
Add to vignette (SPR or SPS)
exploreDDS,exploreDDSplot,GLMplot,MAplot,MDSplot,PCAplot,hclustplot,heatMaplot,tSNEplot,volcanoplot
Enrichment analysis and visualization tool for SPR
- Integration with
FGSEA
WebSite
Updated the vignette
Added systemPipeRdata vignette and presentation: link
Redirect http://girke.bioinformatics.ucr.edu/systemPipeR/ to new page
Add content to FAQ section
Add tutorials videos
class: middle
Thanks!
Ask a question about systemPipeR at Bioconductor Support Page
systemPipeRdata at Bioconductor