Runs DESeq2
run_DESeq2.RdConvenience wrapper function to identify differentially expressed genes
(DEGs) in batch mode with DESeq2 for any number of pairwise sample
comparisons specified under the cmp argument. Users are strongly
encouraged to consult the DESeq2 vignette for more detailed information
on this topic and how to properly run DESeq2 on data sets with more
complex experimental designs.
run_DESeq2(countDF, targets, cmp, independent = FALSE, lfcShrink=FALSE, type="normal")Arguments
- countDF
date.framecontaining raw read counts- targets
targets
data.frame- cmp
character matrixwhere comparisons are defined in two columns. This matrix should be generated with thereadComp()function from the targets file. Values used for comparisons need to match those in theFactorcolumn of the targets file.- independent
If
independent=TRUEthen thecountDFwill be subsetted for each comparison. This behavior can be useful when working with samples from unrelated studies. For samples from the same or comparable studies, the settingindependent=FALSEis usually preferred.- lfcShrink
logiacal. If
TRUEadds shrunken log2 fold changes (LFC) to the object.- type
please check
DESeq2::lfcShrink()documentation. Availablecharacteralternatives: "apeglm"; "ashr"; "normal".
Value
data.frame containing DESeq2 results from all comparisons. Comparison labels are appended to column titles for tracking.
References
Please properly cite the DESeq2 papers when using this function:
http://www.bioconductor.org/packages/devel/bioc/html/DESeq2.html
See also
run_edgeR, readComp and DESeq2 vignette
Examples
targetspath <- system.file("extdata", "targets.txt", package="systemPipeR")
targets <- read.delim(targetspath, comment.char = "#")
cmp <- readComp(file=targetspath, format="matrix", delim="-")
countfile <- system.file("extdata", "countDFeByg.xls", package="systemPipeR")
countDF <- read.delim(countfile, row.names=1)
degseqDF <- run_DESeq2(countDF=countDF, targets=targets, cmp=cmp[[1]], independent=FALSE)
#> Loading required namespace: DESeq2
#> Warning: some variables in design formula are characters, converting to factors
pval <- degseqDF[, grep("_FDR$", colnames(degseqDF)), drop=FALSE]
fold <- degseqDF[, grep("_logFC$", colnames(degseqDF)), drop=FALSE]
DEG_list <- filterDEGs(degDF=degseqDF, filter=c(Fold=2, FDR=10))
 names(DEG_list)
#> [1] "UporDown" "Up" "Down" "Summary"
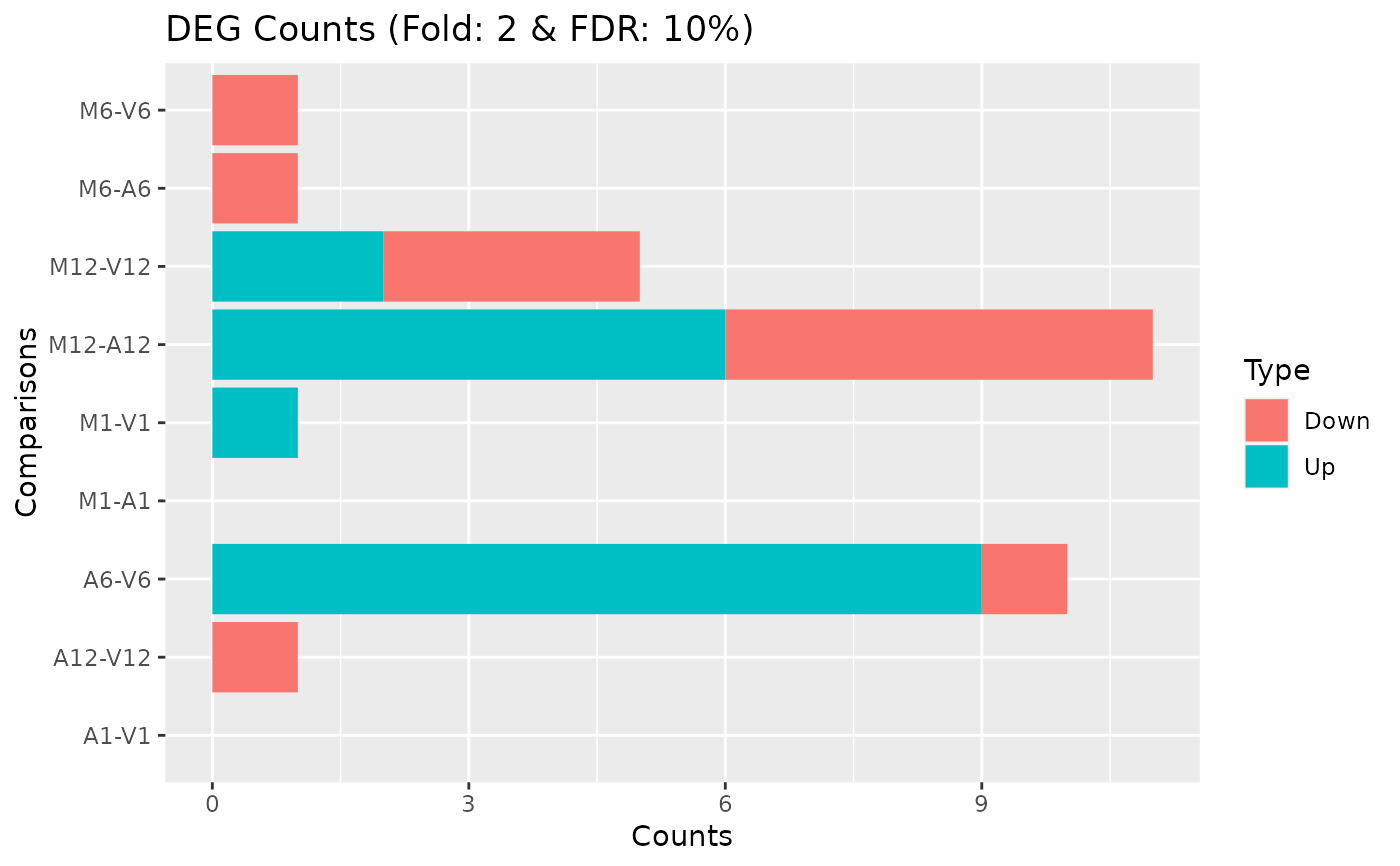
DEG_list$Summary
#> Comparisons Counts_Up_or_Down Counts_Up Counts_Down
#> M1-A1 M1-A1 0 0 0
#> M1-V1 M1-V1 1 1 0
#> A1-V1 A1-V1 0 0 0
#> M6-A6 M6-A6 1 0 1
#> M6-V6 M6-V6 1 0 1
#> A6-V6 A6-V6 10 9 1
#> M12-A12 M12-A12 11 6 5
#> M12-V12 M12-V12 5 2 3
#> A12-V12 A12-V12 1 0 1
names(DEG_list)
#> [1] "UporDown" "Up" "Down" "Summary"
DEG_list$Summary
#> Comparisons Counts_Up_or_Down Counts_Up Counts_Down
#> M1-A1 M1-A1 0 0 0
#> M1-V1 M1-V1 1 1 0
#> A1-V1 A1-V1 0 0 0
#> M6-A6 M6-A6 1 0 1
#> M6-V6 M6-V6 1 0 1
#> A6-V6 A6-V6 10 9 1
#> M12-A12 M12-A12 11 6 5
#> M12-V12 M12-V12 5 2 3
#> A12-V12 A12-V12 1 0 1