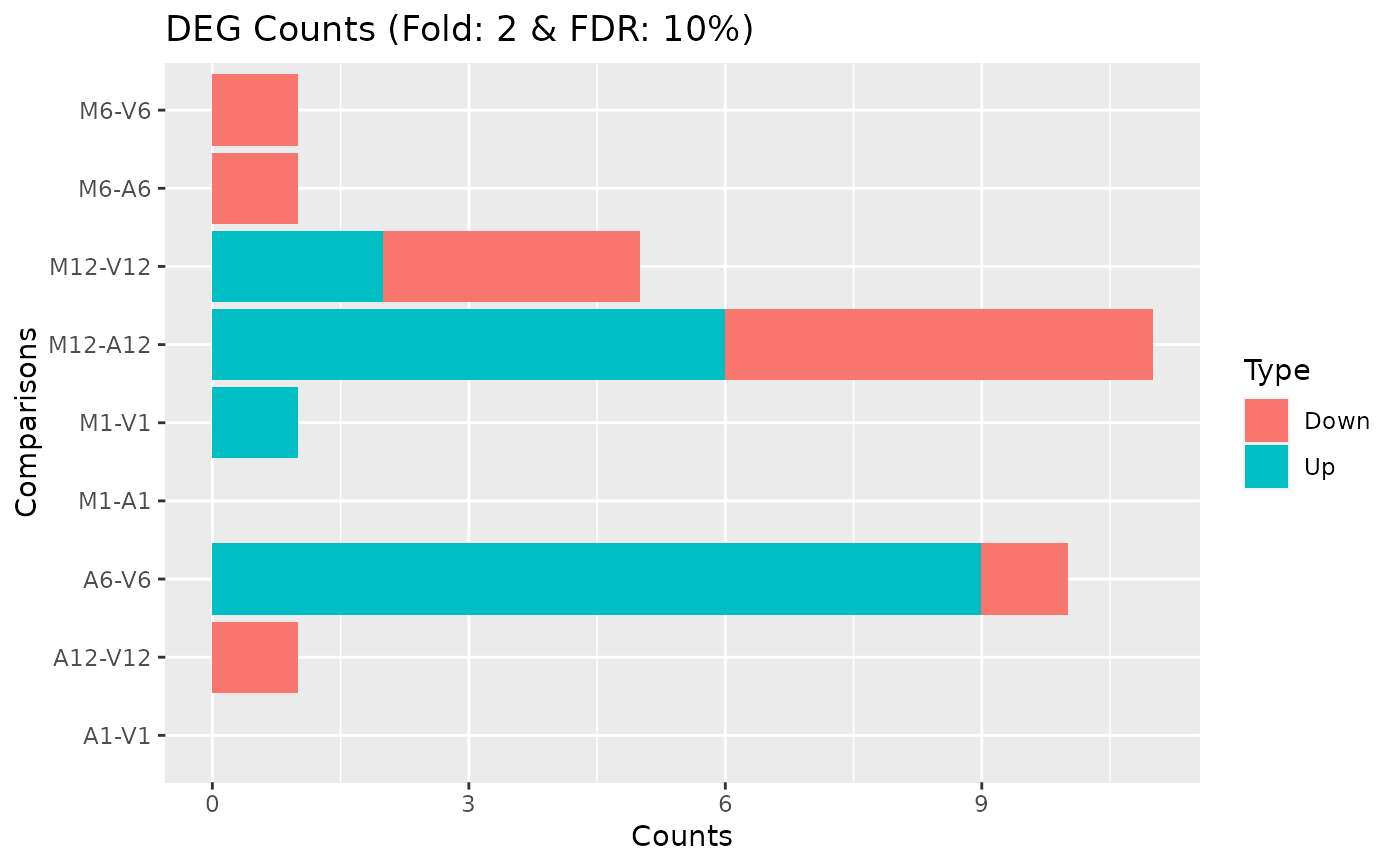
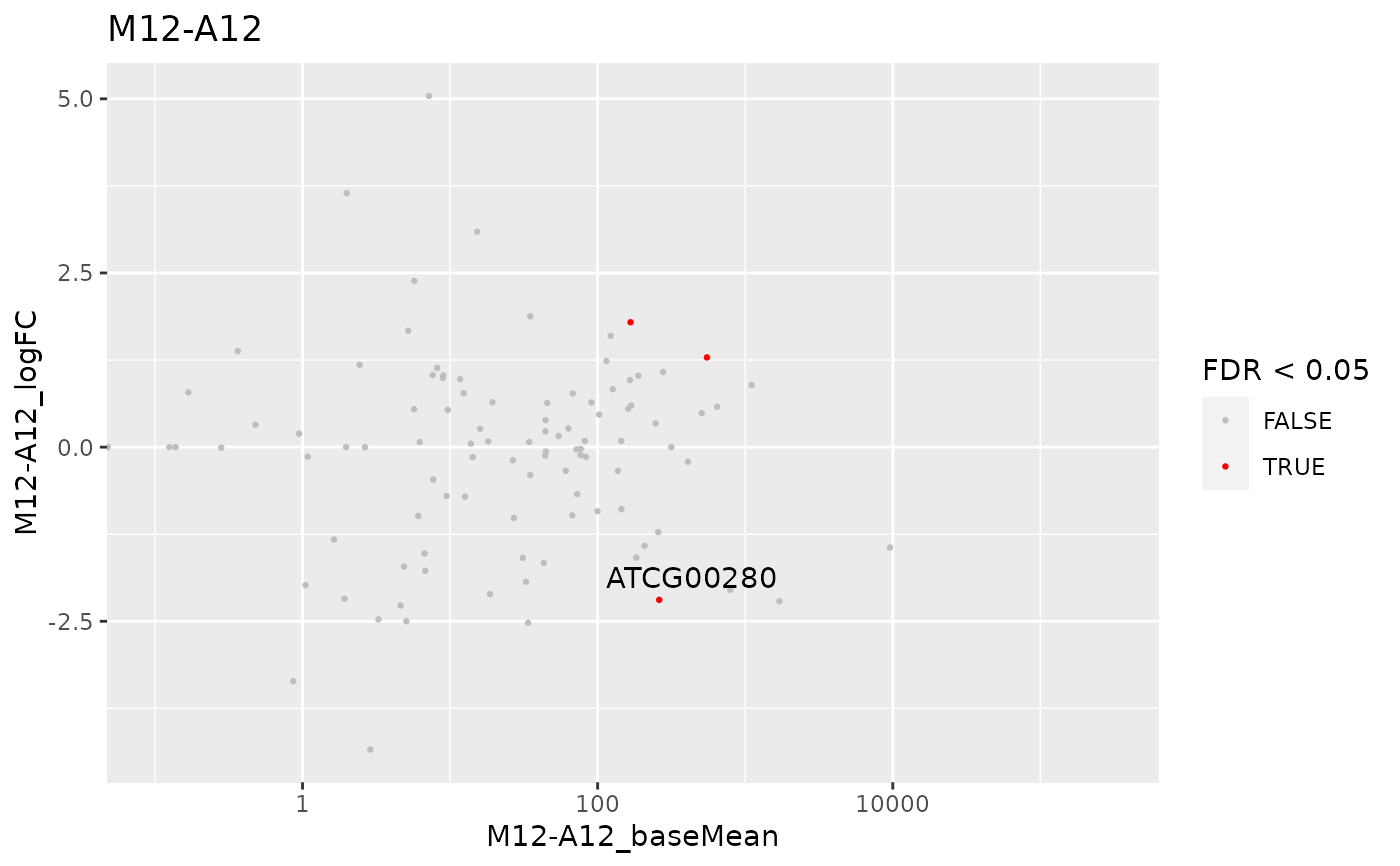
MAplot
MAplot.RdThis function plots log2 fold changes (y-axis) versus the mean of normalized counts (on the x-axis). Statistically significant features are colored.
MAplot( degseqDF, FDR.cutoff = 0.05, comparison, filter = c(Fold = 2, FDR = 10), genes = "NULL", plotly = FALSE, savePlot = FALSE, filePlot = NULL )
Arguments
| degseqDF | object of class |
|---|---|
| FDR.cutoff | filter cutoffs for the p-value adjusted. |
| comparison |
|
| filter | Named vector with filter cutoffs of format c(Fold=2, FDR=1) where Fold refers to the fold change cutoff (unlogged) and FDR to the p-value cutoff. |
| genes |
|
| plotly | logical: when |
| savePlot | logical: when |
| filePlot | file name where the plot will be saved. For more information,
please consult the |
Value
returns an object of ggplot or plotly class.
Examples
## Load targets file and count reads dataframe targetspath <- system.file("extdata", "targets.txt", package = "systemPipeR") targets <- read.delim(targetspath, comment = "#") cmp <- systemPipeR::readComp( file = targetspath, format = "matrix", delim = "-" ) countMatrixPath <- system.file("extdata", "countDFeByg.xls", package = "systemPipeR" ) countMatrix <- read.delim(countMatrixPath, row.names = 1) ### DEG analysis with `systemPipeR` degseqDF <- systemPipeR::run_DESeq2( countDF = countMatrix, targets = targets, cmp = cmp[[1]], independent = FALSE )#> Warning: some variables in design formula are characters, converting to factors## Plot MAplot(degseqDF, comparison = "M12-A12", filter = c(Fold = 1, FDR = 20), genes = "ATCG00280" )